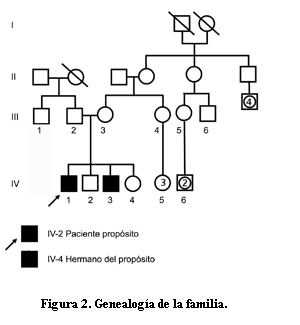
También conocida como drepanocitosis o anemia depranocitica, es una hemoglobinopatía, enfermedad genética autosómica recesiva resultado de la sustitución de adenina por timina en el gen de la globina beta, ubicado en el cromosoma 11, lo que conduce a una mutación de ácido glutámico por valina en la posición de la cadena poli peptídica de globina beta y a la producción de una hemoglobina funcionalmente defectuosa, la hemoglobina S, que afecta la hemoglobina una proteína que forma parte de los glóbulos rojos y se encarga del transporte de oxígeno. Es de origen genético y se da por la sustitución de un aminoácido en su conformación, esto provoca que a baja tensión de oxigeno la hemoglobina se deforme y el eritrocito adquiera apariencia de una hoz, la nueva forma provoca dificultad para la circulación de los glóbulos rojos, por ello se obstruyen los vasos sanguíneos y causan síntomas con dolor en las extremidades. Los glóbulos rojos también padecen de una vida más corta provocando anemia por no ser reemplazados a tiempo.
La anemia falciforme ocurre cuando una persona hereda 2 genes anómalos (uno de cada progenitor), lo que determina que sus glóbulos rojos tengan una forma anómala. En vez de ser flexibles y en forma de disco, son rígidos y curvos, adoptando la forma de una antigua herramienta de labranza denominada hoz de donde proviene el nombre de la enfermedad (falciforme se significa relativo a la hoz), parecida a una luna en cuarto creciente.
La anemia falciforme y su alelo Si para la sangre normal. La condición falciforme se caracteriza por una deficiencia en la hemoglobina que resulta en una forma alterada de los corpúsculos rojos. Un simple par de alelos está involucrado en la anemia falciforme, enfermedad altamente mortal. La anemia falciforme es común en algunas partes de África. Cerca del 40% de las personas en algunas tribus negras llevan el gene. El gene es virtualmente inexistente en otros grupos raciales.
La malaria es común en las mismas áreas en donde predomina la anemia falciforme, pero la mayor incidencia de mortalidad de malaria ocurre entre nativos con hemoglobina normal en sus corpúsculos rojos.
Las personas heterocigotos (Si / Si) son así favorecidas en las áreas en que la malaria predomina; su sangre es resistente al parasito de la malaria y la anemia no es severa. Los homocigotas para la característica falciforme (Si / Si) generalmente mueren pronto de anemia, los homocigóticos para las características normales de hemoglobina (Si / Si) sufren la malaria y los heterocigóticos (Si / Si) viven razonablemente sanos.
Si la malaria fuese controlada, el polimorfismo desaparecía y la población seria esencialmente homocigótica para Sia, excepto para mutaciones raras de Sia a Si s.
Otra enfermedad de la hemoglobina, algo similar a la anemia falciforme, es la talasemia (o anemia de Cooley) que ocurre en su mayor parte de niños y es casi 100% fatal. La enfermedad está controlada por un simple gene en condición homocigótica (cc), produce la anemia severa de Cooley o talasemia mayor. El mismo gene en condición heterocigótica resulta en una forma benigna de la enfermedad llamada talasemia menor o microcitemia. Las personas con la combinación heterocigótica no presentan síntomas.
El gene de la talasemia se extiende a la mayor parte de las áreas ecuatoriales de la tierra. En los Estados Unidos, el gene es raro. El gene especialmente frecuente en la región mediterránea particularmente en Italia, Grecia y Siria. En algunas áreas locales pueden presentarse el 10% de portadores. En áreas en donde los portadores se presentan en frecuencias de 1 en 20, la probabilidad de la forma severa (que requiere la condición homocigótica) que ocurre por los cruzamientos al azar es de alrededor de 1 en 400. La forma homocigótica es letal y la heterocigótica puede ser identificada temprano en la vida.
Las variaciones de los grupos sanguíneos son las características humanas mejor conocidas y han sido muy útiles sugerir conexiones ancestrales y mecanismos involucrados normales tienen forma de disco y son muy flexibles. En la enfermedad de células falciformes, algunos glóbulos rojos pueden hacerse más rígidos y cambiar de forma de modo tal que toma forma de hoz o de una luna creciente. Estos no se pueden mover muy bien a través de los vasos sanguíneos más pequeños. Esto puede interrumpir o disminuir el flujo de sangre a ciertas partes del cuerpo haciendo que llegue menos oxígeno a estas áreas. Las células depranocitica también mueren antes que los glóbulos rojos normales y esto puede causar una disminución de los glóbulos rojos en el cuerpo. No existe cura para la enfermedad de células falciformes en la mayoría de las personas.
Las hemoglobinopatías especialmente las talasemias y la anemia falciforme, están extendidas por todo el mundo. Cerca del 5 % de la población mundial es portadora de genes causantes de hemoglobinopatías. Cada año nacen aproximadamente 300 000 niños con hemoglobinopatías importantes, de los cuales más de 200 000 son africanos con anemia falciforme.
La anemia depranocitica es especialmente frecuente en personas con antepasados originarios del África subsahariana, la India, la Arabia Saudita o los países del Mediterráneo. En algunas zonas del África subsahariana, el porcentaje de niños con este trastorno puede llegar al 2%. La prevalencia de la anemia falciforme al nacer depende de la frecuencia del estado de portador. La anemia falciforme tiene importantes repercusiones de salud pública. Sus efectos en la salud humana se pueden evaluar en función de la mortalidad infantil y de niños menores de cinco años. Como no todas las muertes se producen en el primer año de vida, la medida más valida es la mortalidad de los menores de cinco años. La proporción de niños afectados que sobreviven más allá de los cinco años es cada vez mayor, pero esos niños corren el riesgo de muerte prematura. Cuando el impacto en la salud se mide en función de la mortalidad de los menores de cinco años, la anemia falciforme es la causa de la muerte de un 5% de este segmento de la población en el continente africano, de más de un 90% en África occidental y de hasta un 16% en algunos países de esta región.
La anemia falciforme tiene un amplio espectro clínico. La mayoría de los afectados que tienen anemia crónica con una hemoglobinemia de alrededor de 8 g/dl. Los principales problemas se deben a la tendencia de los eritrocitos a adoptar una morfología falciforme y a bloquear los capilares cuando la tensión del oxígeno es baja. En los niños , los eritrocitos falciformes tienden a quedar atrapados en el bazo, lo cual ocasiona un serio riesgo de muerte antes de los siete años por crisis súbitas de anemia profunda asociadas a la rápida esplenomegalia, o por el hipoesplenismo, que permite que se produzcan infecciones muy graves. Entre los 6 y los 18 meses de vida, os niños afectados suelen presentar tumefacción dolorosa de las manos o los pies (síndrome de mano-pie). Los supervivientes también pueden sufrir crisis dolorosas graves, recurrentes e impredecibles, así como “síndrome acido agudo” (neumonía o infarto pulmonar), necrosis ósea o articular, priapismo o insuficiencia renal.
Referencias
Rund, Pand Rachmilewitz, Medical Progress: Beta Thalassemia, New England Journalist of Medicine, Volumen 353, N° 11,15 de setiembre de 2005, pag.1135-1146.